Sequence logos for DNA sequence alignments
Oliver Bembom
Division of Biostatistics, University of California, BerkeleyRobert Ivánek
Department of Biomedicine, University of Basel, Basel, SwitzerlandSwiss Institute of Bioinformatics, Basel, Switzerlandrobert.ivanek@unibas.ch Source:
vignettes/seqLogo.Rmd
seqLogo.RmdLast edited: 08 May 2020
Introduction
An alignment of DNA or amino acid sequences is commonly represented in the form of a position weight matrix (PWM), a \(J \times W\) matrix in which position \((j,w)\) gives the probability of observing nucleotide \(j\) in position \(w\) of an alignment of length \(W\). Here \(J\) denotes the number of letters in the alphabet from which the sequences were derived. An important summary measure of a given position weight matrix is its information content profile (Schneider et al. 1986). The information content at position \(w\) of the motif is given by
\[ IC(w) = \log_2(J) + \sum_{j=1}^J p_{wj}\log_2(p_{wj}) = \log_2(J) - entropy(w). \]
The information content is measured in bits and, in the case of DNA sequences, ranges from 0 to 2 bits. A position in the motif at which all nucleotides occur with equal probability has an information content of 0 bits, while a position at which only a single nucleotide can occur has an information content of 2 bits. The information content at a given position can therefore be thought of as giving a measure of the tolerance for substitutions in that position: Positions that are highly conserved and thus have a low tolerance for substitutions correspond to high information content, while positions with a high tolerance for substitutions correspond to low information content.
Sequence logos are a graphical representation of sequence alignments developed by (Schneider and Stephens 1990). Each logo consists of stacks of symbols, one stack for each position in the sequence. The overall height of the stack is proportional to the information content at that position, while the height of symbols within the stack indicates the relative frequency of each amino or nucleic acid at that position. In general, a sequence logo provides a richer and more precise description of, for example, a binding site, than would a consensus sequence.
Software implementation
The seqLogo
package provides an R implementation for plotting such sequence logos
for alignments consisting of DNA sequences. Before being able to access
this functionality, the user is required to load the package using the
library() command:
library(seqLogo) The pwm-class
The seqLogo
package defines the class pwm which can be used to
represent position weight matrices. An instance of this class can be
constructed from a simple matrix or a data frame using the function
makePWM():
mFile <- system.file("extdata/pwm1", package="seqLogo")
m <- read.table(mFile)
m
## V1 V2 V3 V4 V5 V6 V7 V8
## 1 0.0 0.0 0.0 0.3 0.2 0.0 0.0 0.0
## 2 0.8 0.2 0.8 0.3 0.4 0.2 0.8 0.2
## 3 0.2 0.8 0.2 0.4 0.3 0.8 0.2 0.8
## 4 0.0 0.0 0.0 0.0 0.1 0.0 0.0 0.0
p <- makePWM(m)makePWM() checks that all column probabilities add up to
1.0 and also obtains the information content profile and consensus
sequence for the position weight matrix. These can then be accessed
through the corresponding slots of the created object:
slotNames(p)
## [1] "pwm" "width" "ic" "alphabet" "consensus"
pwm(p)
## 1 2 3 4 5 6 7 8
## A 0.0 0.0 0.0 0.3 0.2 0.0 0.0 0.0
## C 0.8 0.2 0.8 0.3 0.4 0.2 0.8 0.2
## G 0.2 0.8 0.2 0.4 0.3 0.8 0.2 0.8
## T 0.0 0.0 0.0 0.0 0.1 0.0 0.0 0.0
ic(p)
## [1] 1.2780719 1.2780719 1.2780719 0.4290494 0.1535607 1.2780719 1.2780719
## [8] 1.2780719
consensus(p)
## [1] "CGCGCGCG"Plotting sequence logos
The seqLogo() function plots sequence logos.
Input
The position weight matrix for which the sequence logo is to be plotted,
pwm. This may be either an instance of classpwm, as defined by the package seqLogo, amatrix, or adata.frame.A
logicalic.scaleindicating whether the height of each column is to be proportional to its information content, as originally proposed by (Schneider et al. 1986). Ific.scale=FALSE, all columns have the same height.
Example
The call seqLogo(p) produces the sequence logo shown in
figure @ref(seqlogo1). Alternatively, we can use
seqLogo(p, ic.scale=FALSE) to obtain the sequence logo
shown in figure @ref(seqlogo2) in which all columns have the same
height.
seqLogo(p)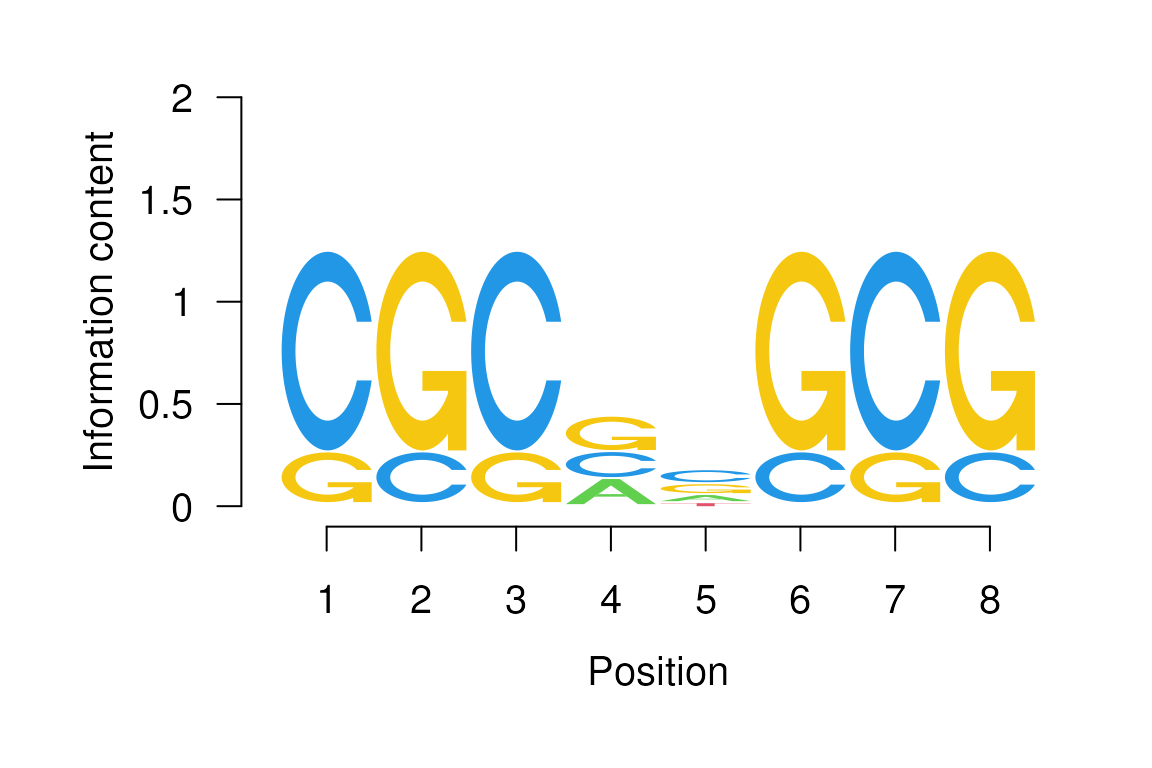
Sequence logo with column heights proportional to information content.
seqLogo(p, ic.scale=FALSE)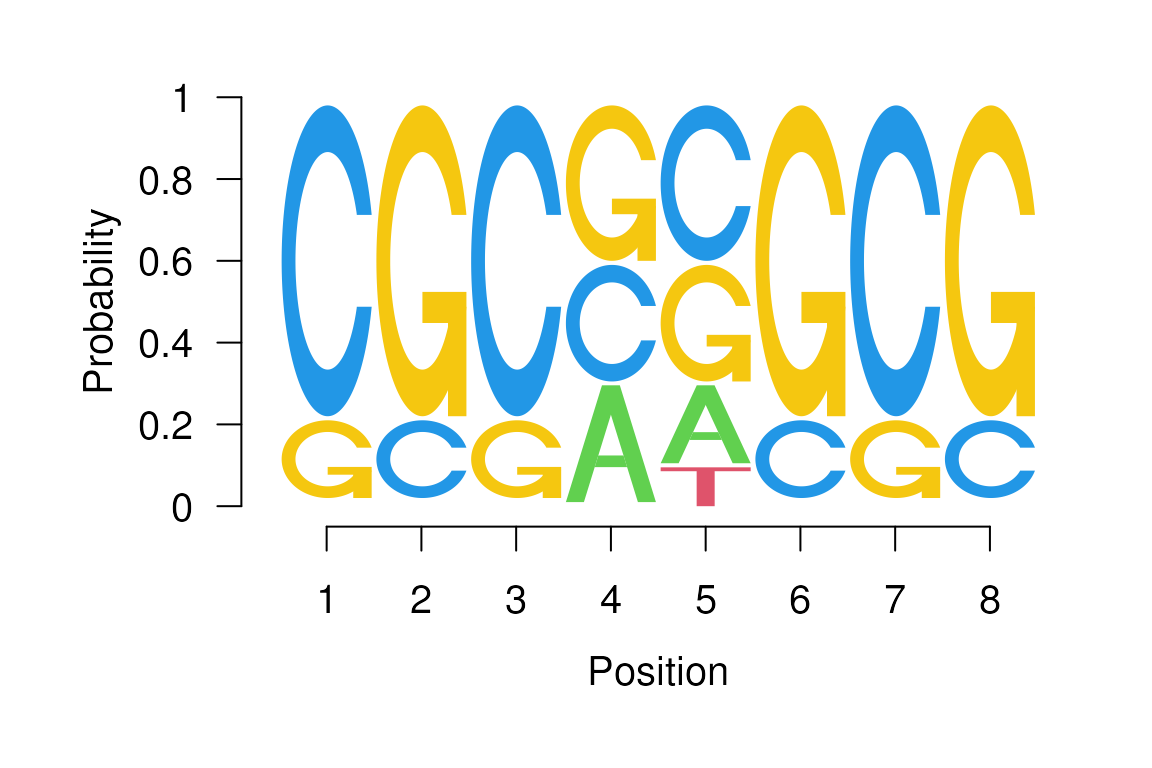
Sequence logo with uniform column heights.
It is also possible to change the default colors by providing a named
character vector as a fill argument seqLogo
function.
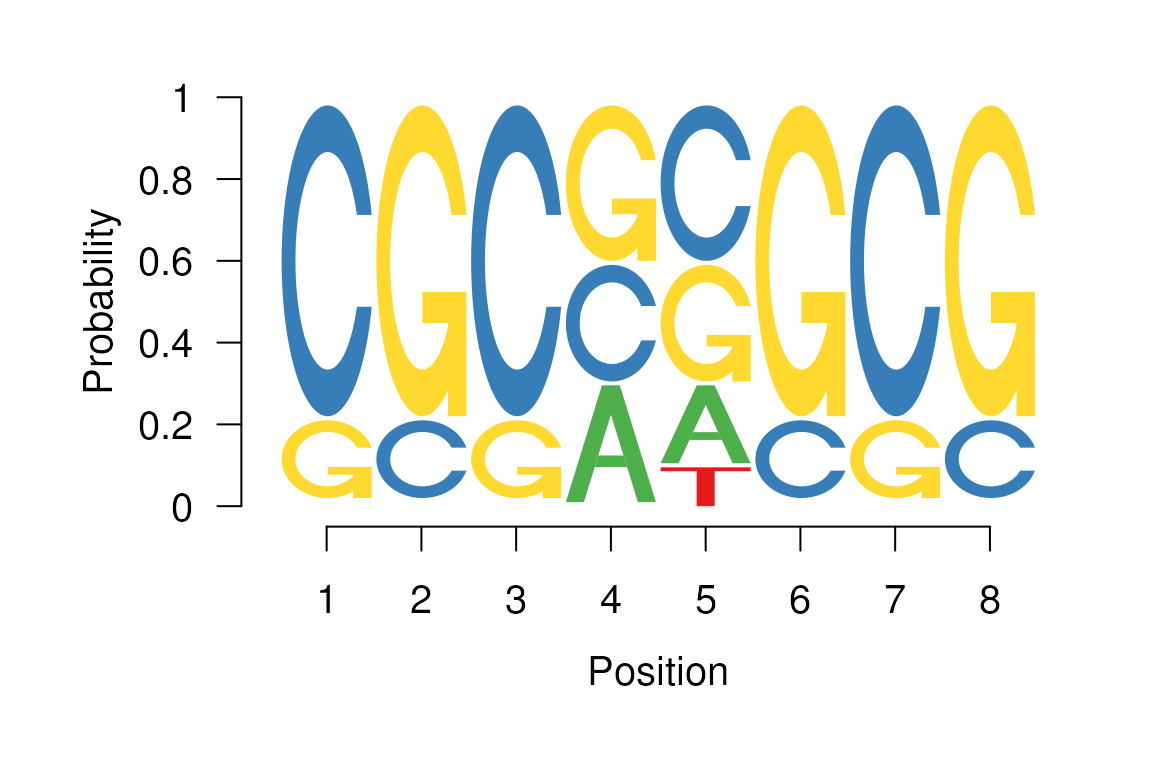
Sequence logo with user specified colors.
The RNA logos are supported as well. In this particular case, the
seqLogo will either accept fill colors
specified for c("A", "C", "G", "U") letters or
c("A", "C", "G", "T") and uses the color specified in
element “T” for letter “U”.
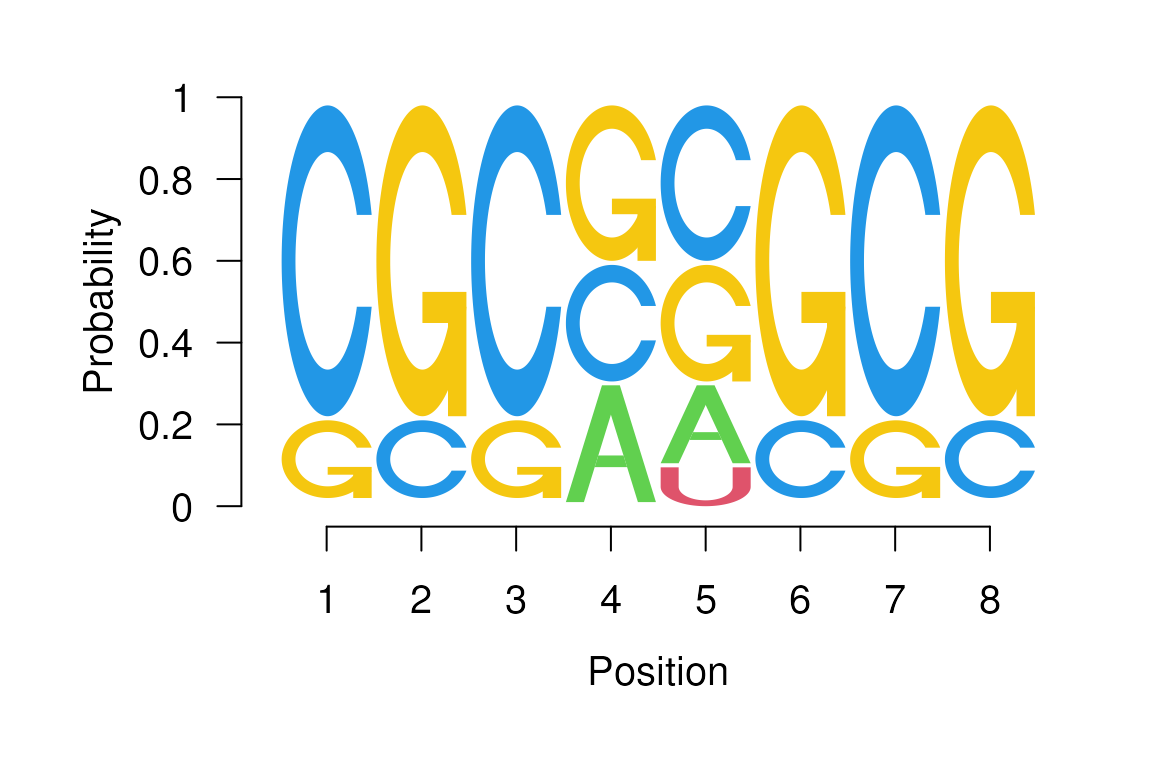
RNA Sequence logo.
Software Design
The following features of the programming approach employed in seqLogo may be of interest to users.
Class/method object-oriented programming. Like many
other Bioconductor packages, seqLogo
has adopted the S4 class/method objected-oriented programming
approach presented in (Chambers
1998). In particular, a new class, pwm, is defined
to represent a position weight matrix. The plot method for this class is
set to produce the sequence logo corresponding to this class.
Use of the grid package. The
grid package is used to draw the sequence letters from
graphical primitives. We note that this should make it easy to extend
the package to amino acid sequences.
SessionInfo
The following is the session info that generated this vignette:
sessionInfo()
## R version 4.3.0 (2023-04-21)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.2 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] grid stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] seqLogo_1.67.0 BiocStyle_2.29.0
##
## loaded via a namespace (and not attached):
## [1] vctrs_0.6.2 cli_3.6.1 knitr_1.42
## [4] rlang_1.1.1 xfun_0.39 highr_0.10
## [7] stringi_1.7.12 purrr_1.0.1 textshaping_0.3.6
## [10] jsonlite_1.8.4 glue_1.6.2 rprojroot_2.0.3
## [13] htmltools_0.5.5 stats4_4.3.0 ragg_1.2.5
## [16] sass_0.4.5 rmarkdown_2.21 evaluate_0.20
## [19] jquerylib_0.1.4 fastmap_1.1.1 lifecycle_1.0.3
## [22] yaml_2.3.7 memoise_2.0.1 bookdown_0.33
## [25] BiocManager_1.30.20 stringr_1.5.0 compiler_4.3.0
## [28] fs_1.6.2 systemfonts_1.0.4 digest_0.6.31
## [31] R6_2.5.1 magrittr_2.0.3 bslib_0.4.2
## [34] tools_4.3.0 pkgdown_2.0.7.9000 cachem_1.0.8
## [37] desc_1.4.2